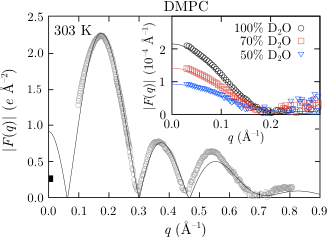
X-ray and neutron scattering form factors obtained from simulations with our force field and compared to experimental data.

Verification of Stockholm lipids
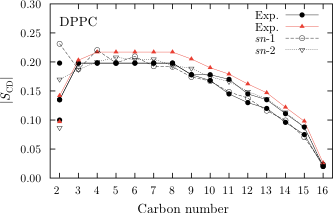
NMR deuterium order parameters for DPPC
Free energy of transfer of amino acid analogues from water to cyclohexane. Parameters for the amino acids analogues are taken from the AMBER03 force field.

When verifying Stockholm lipids we focused on seeing if the force field could reproduce experimental raw data, i.e. data that is free from assumptions etc.
This means that we looked mostly at scattering form factors from X-ray/neutron scattering studies and deuterium order parameters from NMR studies.
Since Stockholm lipids can reproduce these very well it is safe to say that all the other structural data obtained from this force field is accurate.
This means that the force field available under Downloads is very suitable for simulations of biological membranes in a tensionless ensemble, i.e. NPT, which is the proper ensemble for membrane simulations.
In order to be able to simulate membrane bound proteins in their native environment Stockholm lipids has been developed according to the AMBER force field for amino acids. We have performed simulations with explicit membrane proteins and also computed free energies of transfer between water and cyclohexane (modeled with the Lennard-Jones parameters used for the lipid tails).
See ref. 1 and ref. 2 from the Publications section for the complete validation