Distrubutions of Counterions around DNA. Molecular Dynamics Simulations Results.
Alexander Lyubartsev and Aatto LaaksonenSupplementary matherial to the paper: A.P.Lyubartsev, A.Laaksonen, "Molecular Dynamics Simulation of DNA in Presence of Different Counterions" Journal of Biomolecular Structure and Dynamics, v.16. p.579 (1998)
Introduction
We have carried out a series of molecular dynamics simulations of DNA (see details below) in presence of water and three types of counterions: Li, Na and Cs. The results on spatial distributions of ions, diffusion of ions near the DNA surface and on effective (solvent-mediated) ion-DNA interaction potentials will be published soon.This page contains supplementary material concerning
mainly spatial distribution functions (SDF). The spatial distribution function
is a 3-dimensional distribution of a molecule (or of an atom of a molecule)
in local coordinate system of another molecule. The presentation of spatial
distribution functions is tricky. The full representation requires
four-dimensional space (three space coordinates plus one for the intensity).
The standard way to proceed is to use 3D graphics and display iso-intensity
surfaces. The GOpenMol package by Leif Laaksonen provides a nice
tool to do this. You may both rotate the 3D picture and change the
intensity level interactilely. The best way to view the spatial distribution
function is to have GOpenMol installed as a helper application of your
Web browser. See below instructions how to get
and install GOpenMol. Some animated pictures of SDF (the intensity
level changes with time) are also presented on this page.
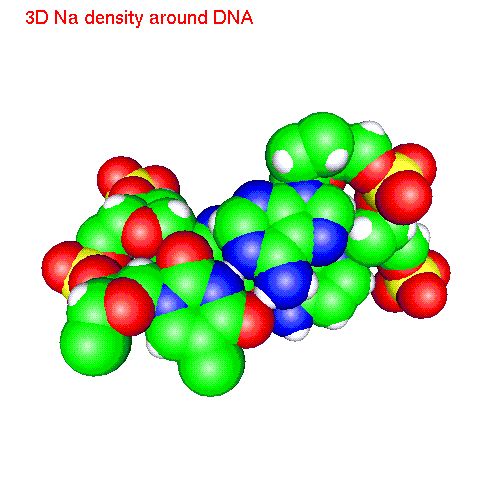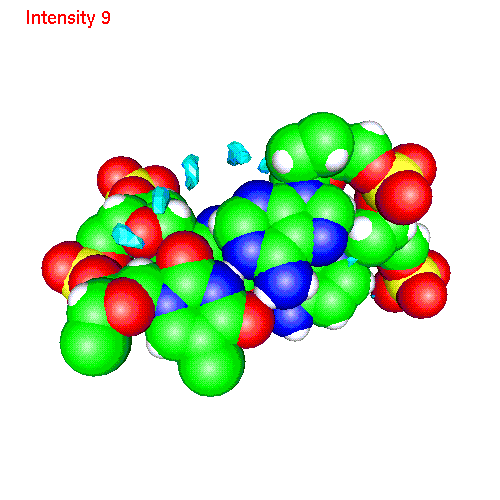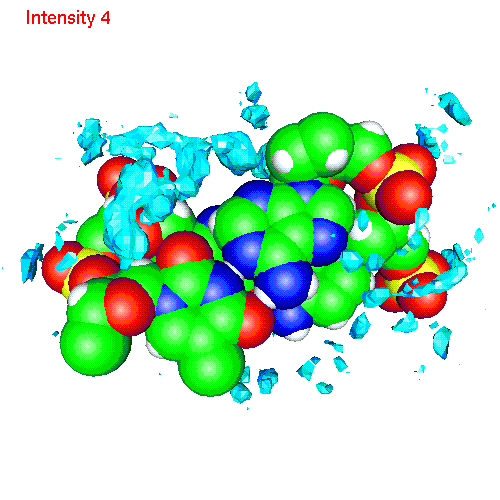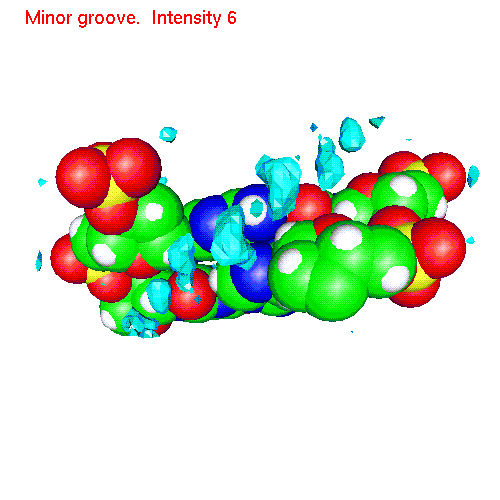
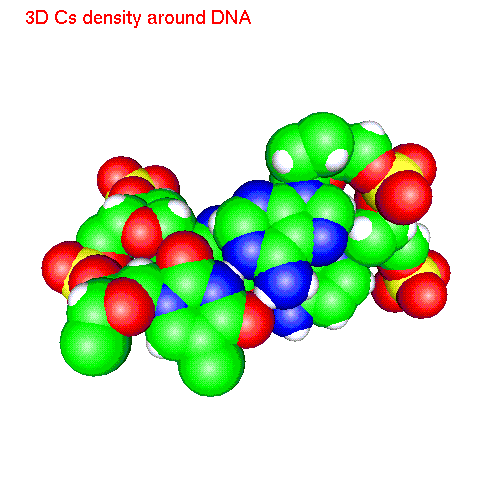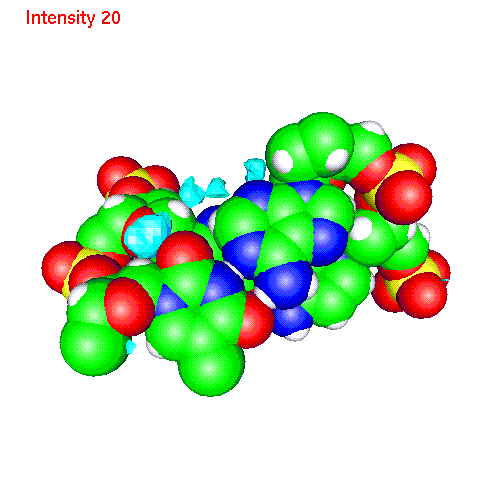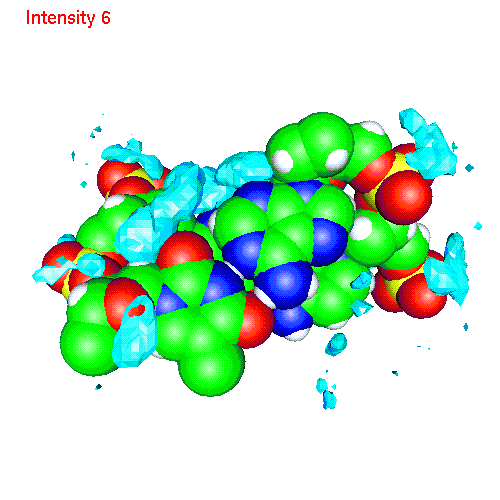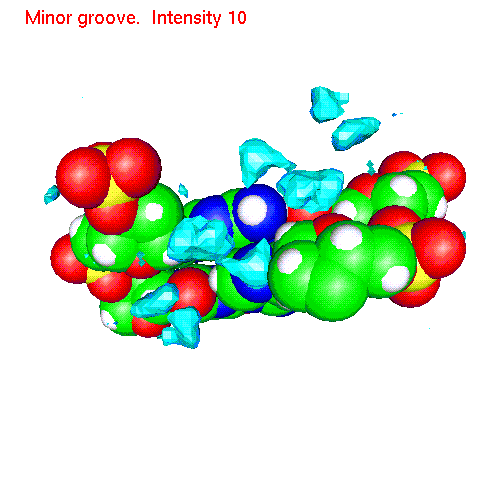
Spatial distribution functions of ions around DNA
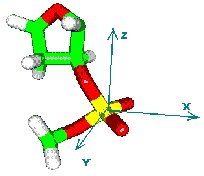
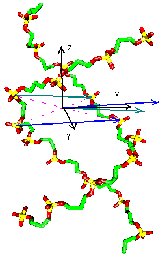 To build
a spatial distribution function one should fix the local coordinate system.
For large molecules it may be done in different ways. Picture to the right
explains how we fix the local coordinate system using four phosphates of
two neighbouring base pairs.
To build
a spatial distribution function one should fix the local coordinate system.
For large molecules it may be done in different ways. Picture to the right
explains how we fix the local coordinate system using four phosphates of
two neighbouring base pairs.
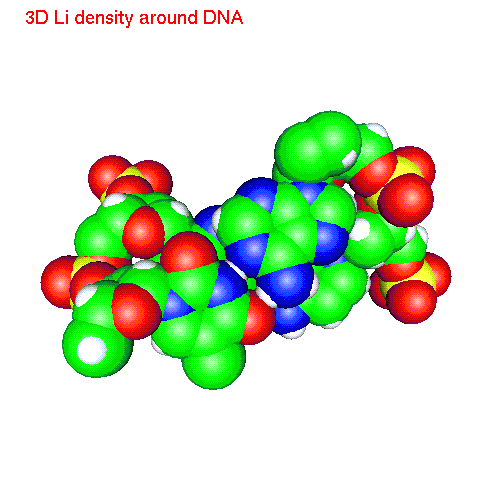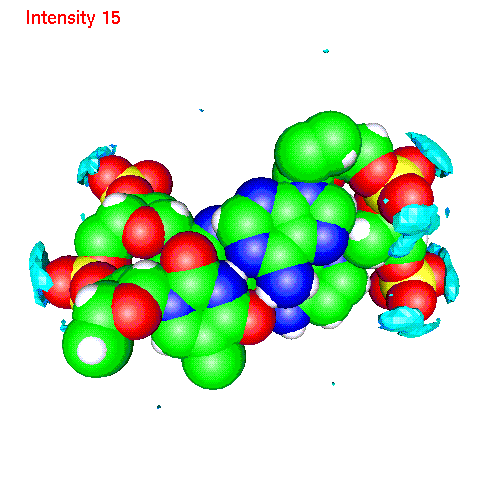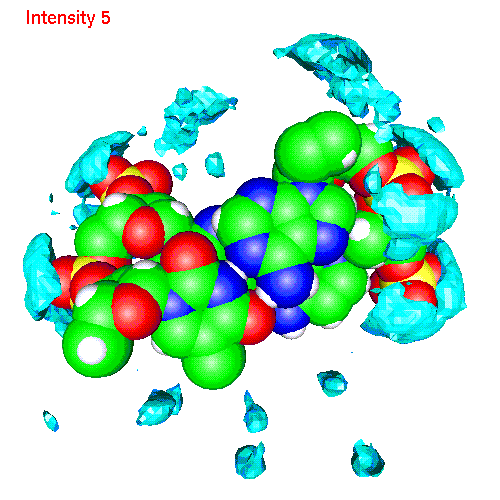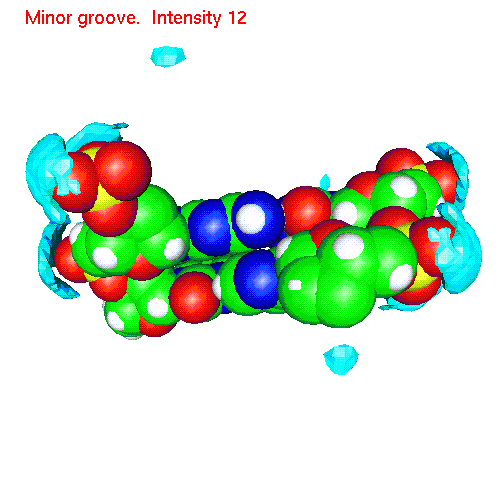
One can also calculate average positions of atoms of the molecule in
the local coordinate system and obtain in this way "averaged molecular
structure". On the pictures below we displayed the averaged structures
of a two base pairs fragment and spatial distribution functions of
ions around it. As an example we displayed d(AC)d(TG) fragment, while the
displayed spatial distribution functions were obtained by averaging over
the all 20 pairs (10 in each direction) of our sample.
Spatial distribution of ions around two base pairs fragment:
| Counterions | Gopenmol files | Animated SDF |
| Li | baseLi.gom (1.4Mb) | LiSDF.gif (480 K) |
| Na | baseNa.gom (1.2Mb) | NaSDF.gif (475 K) |
| Cs | baseCs.gom (1.4Mb) | CsSDF.gif (433 K) |
{kind=link}
{kind=link}
{kind=link}
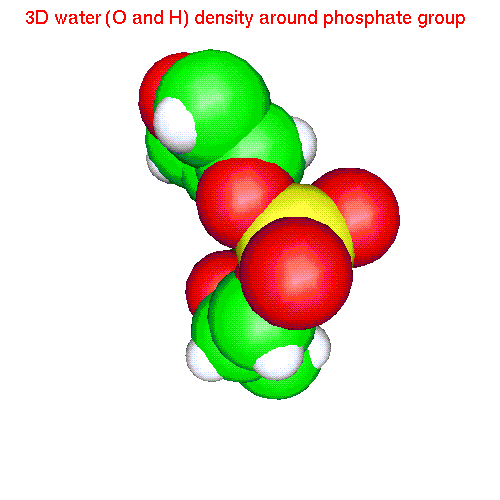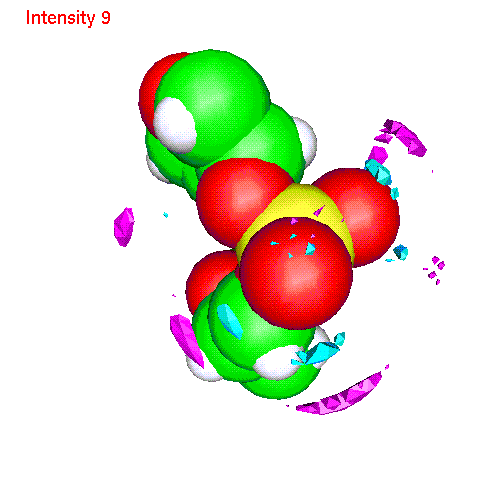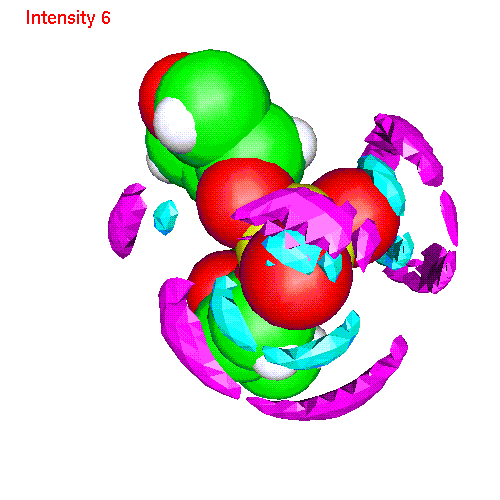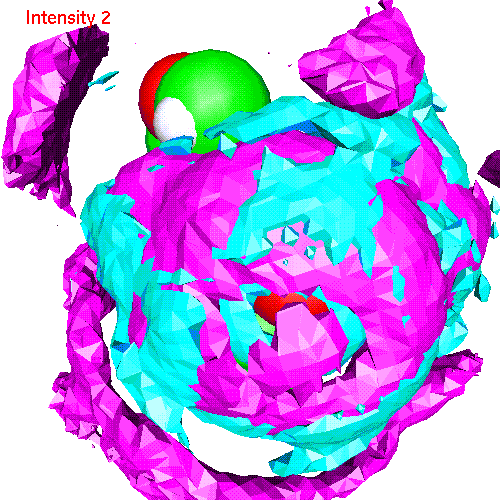
Spatial distributions around a phosphate group
 The local coordinate
system is fixed in this case by phosphorus and two free oxygens:
The local coordinate
system is fixed in this case by phosphorus and two free oxygens:
- Water h2oP.gom(1.2 Mb) Separate isosurfaces for O (red) and H (blue) around phosphate. Animated SDF H2O_SDF.gif (280 K)
- Li ionsLiP.gom (1.2 Mb) Li ions distribution around phosphate. Animated SDF LiP_SDF.gif (327 K)
{kind=link}
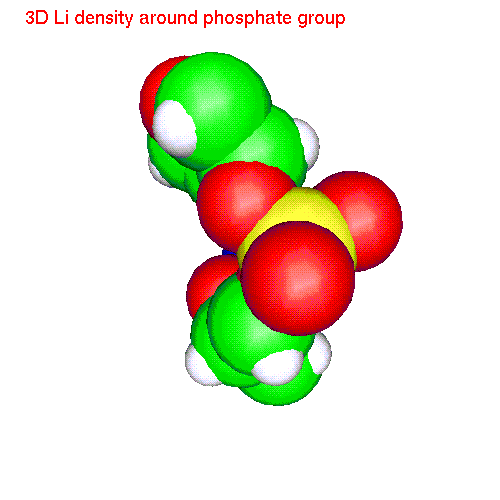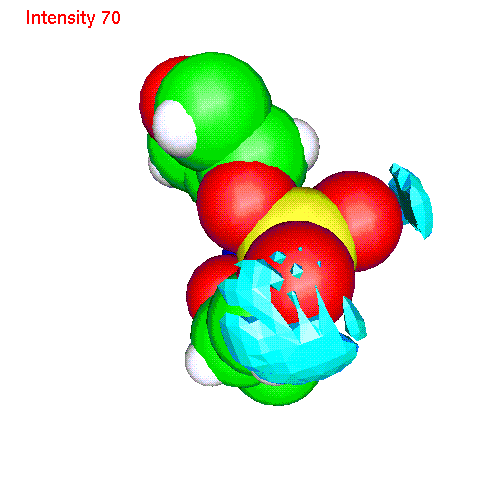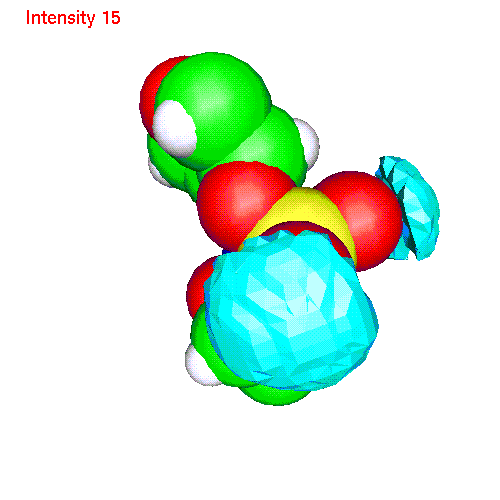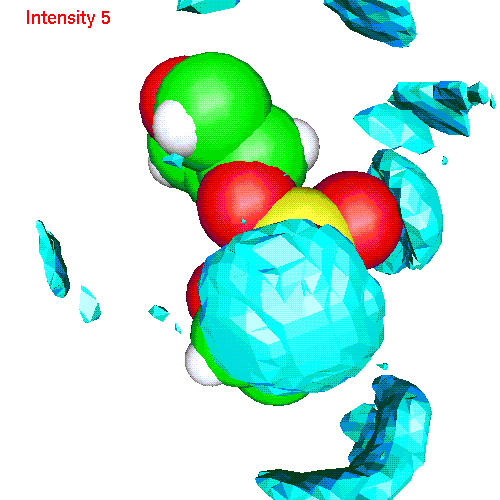{kind=link}
Get and setup GOpenMol
The GOpenMol package for Windows 95/98/NT as well as for a large variety of UNIX platforms (DEC, IBM, Linux, SGI) is freely available from http://www.csc.fi./gopenmol/Installation is easy. Normally one need only to
download the file corresponding your platform, unpack it and run Install
script.
After completing the installation setup GOpenMol as a helper
application for your Web browser. Consult your browser manual (mime-type
chemical/x-gom, extension .gom), or find some hints
here.
If your have installed GOpenMol but were not successful in setting
up it as a helper application, you can download
the files with spatial distribution functions onto your local disk and
then open them with GOpenMol program.