


Next: Example: magic.inp
Up: MagiC core: Inverse Solver
Previous: Input/Output files
Contents
magic.inp: main input file
In the MagiC-core input file (version 2.x) parameters are specified as a set of keywords
ParameterName = Value
and have self-explanatory names (old names from version 1.x are also supported).
Warnings or error messages are issued for missing compulsory parameters or inconsistent values.
Lists of variables (vectors) should be comma separated (i.e. BOX = X, Y, Z).
Comment lines denoted by ! or # are supported as well as empty lines.
For logical parameters most possible acronyms, such as F, .F., False, FALSE are accepted.
For convenience the parameters are divided into five groups. There is however no need to follow
this order, the program accept parameters in any order they follow. The old names of parameters
(which is still possible to use) are given in the brackets.
The following parameters can be defined. The brakets denote alternative keyword
corresponding to v. 1.x. Keywords marked with * are mandatory.
- [itemsep=0mm]
- System parameters:
-
- NMType*
- [NTYP] Number of different molecule types (species) present in the system.
- NameMType*
- [NAMOL] Names of the molecule types present in the system, separated by comma.
Every molecule type should have a respective description file(.mcm).
For example
NameMolType = H2O, DMPC defines 2 names: H2O-for the first molecule type,
and DMPC for the second one. The description files should be named H2O.mcm and
DMPC.mcm.
- NMolMType*
- [NSPEC] Number of molecules of each type, written as a comma-separated list,
e.g.
NMolMType=392,3 defines system, which consists of 392 molecules of the first type,
and 3 molecules of the second type.
- LMoveMType
- [LMOVE] Which molecular types are allowed to move in the Monte Carlo simulation.
List of comma-separated logical values.
Default
LMOVE = True,.., True , i.e. all molecules are allowed to move. Frozen molecules
coordinates has to be specified in a *.xmol file given in InputFrozenCoords parameter.
- Epsilon
- [EPS] Dielectric permittivity constant defining electrostatic interactions in the
system. Default: 1.0
- TEMP*
- Temperature of the system, K
- Box*
- [BOXL,BOYL,BOZL] Periodic cell dimensions in Å, separated by comma. The software
uses rectangular periodic boundary conditions.
- Monte Carlo parameters:
-
- MCSteps*
- [NMKS] Total number of Monte Carlo steps to be performed on every iteration
(including equilibration).
- MCStepsEquil*
- [NMKS0] Number of Monte Carlo steps to be performed for the equilibration.
- MCStepAtom*
- [DR] Maximum displacement in a Monte Carlo single atom displacement step, Å.
To use non-uniform MC step (specific to molecule type), one can provide few values: one per molecule type. Default: 1.0.
- MCStepTransMol
- [MCTRANSSTEP] Maximum displacement in a MC translation of a whole
molecule, Å. To use non-uniform MC step (specific to molecule type), one can provide few values: one per molecule type.
Default: 0.0
- MCStepRotMol
- [MCROTSTEP] Maximum degree of MC rotation of the whole molecule, deg. To use non-uniform MC step (specific to molecule type), one can provide few values: one per molecule type. Default 0.0
- iMCStepTransMol
- [ITRANS] How often (in terms of MC steps) to perform random translation of
a randomly chosen molecule. Default: 0, i.e. never
- iMCStepRotMol
- [IROT] How often (in terms of MC step) to perform random rotation of a randomly
chosen molecule. Default: 0, i.e. never
- iCalcEnergy*
- [IOUT] How often to recalculate the total energy and write energies
and pressure to the log-file. If the difference in total energy before and after the recalculation
is larger than
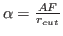 , a warning message will be given. This procedure is computationaly
expensive (order
, a warning message will be given. This procedure is computationaly
expensive (order 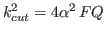 ), and should not be performed too often.
), and should not be performed too often.
- RCutEl*
- [RECUT] Real space cutoff for electrostatic energy in real-space part of the Ewald sum.
It is recommended to set it equal to cutoff radius of RDFs and short-range interactions, but
in some cases other choices can be reasonable.
- AF, FQ
- Ewald summation parameters: The electrostatic energy in Ewald method can be expressed as
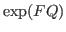 |
(1) |
where,
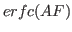 , and
, and
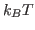 . In other words, the precision or the
first sum is defined by
. In other words, the precision or the
first sum is defined by  , while accuracy of the third sum is defined by
, while accuracy of the third sum is defined by  .
Default:
.
Default: AF=3., FQ=9.0
- RandomSeed
- [NRS] Initial seed for the random number generator.
- KeepStructure
- [LCRDPass] Define if the final structure of the previous inverse iteration
shall be used as starting for the consequent iteration. Default: False
- NMCAutoAdjust
- How many auto adjustments of MCstep-size to perform during equilibration phase? Default: 0 (no autoadjustment)
- MCStepAtomAR
- Desired acceptance ratio for MC atom displacement step (can be either one value, or individual value for every molecule type). Default: 0.5
- MCStepTransMolAR
- Desired acceptance ratio for MC molecule translation step (can be either one value, or individual value for every molecule type). Default: 0.5
- MCStepRotMolAR
- Desired acceptance ratio for MC molecule rotation step (can be either one value, or individual value for every molecule type).
Default: 0.5
- Inverse procedure parameters:
- UseIMC
- [LIMC] Inverse solver selection. If true, the Inverse Monte Carlo method is used,
otherwise iterative Boltzmann inversion is used. Default: True (IMC)
- NIter*
- [IREPT] Number of inverse iterations to perform. Default: 1
- RegP
- [REGP] Regularization parameter for potential correction. This parameter defines
the relative weight of correction, and has a value between 0 and 1. In case of instability
(each next iteration returns larger deviation from reference RDF), value of REGP should be decreased.
It is also advisable if correction to potential at each iteration exceeds treshold value
(default 2
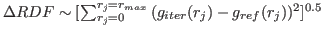 ). REGP can be again increased closer to 1 if the iteration process is stable
and correction to the potential at each iteration is small. Default: 1.0
). REGP can be again increased closer to 1 if the iteration process is stable
and correction to the potential at each iteration is small. Default: 1.0
- iAverage*
- [IAV] How often to compute averages over the system. Since computation of
the averages (RDFs and cross-correlations) involves calculation of distances between all
pairs of atom, this procedure is rather expensive, and should not be performed too often.
The recommended value is of the order of number of CG atoms in the system
The averaging starts after the equilibration, i.e. when first MCStepsEquil steps have passed.
- MaxPotCor
- [DPOTM] Maximal change of potential value at every point during correction
procedure, given in
units. Default: 2.0
- MaxRelDif
- [RTM] Parameter limiting maximum relative difference between reference and
resulting averages. Default: 10.0
- iPotCorrCheck
- How often to perform potential correction check. The program gathers
accumulated statistics from all the processes, and then calculates sampled distribution
functions and potential corrections. However, this corrections are not applied to the actual
interaction potential, but just printed to the log file. This allows user to analyze how well
both distribution functions and potential corrections are converged after given number of MC steps
of an inverse iteration. The checks are performed after equilibration. Default 0, i.e. no check at all.
- ProhibPotLevel
- [POTCUT] Prohibiting potential level. Relatively high value of potential
in
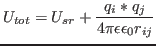 to define a core region of the potentials at distances where the corresponding RDFs are zero,
to avoid MC steps leading to such distances. Default: 1000.
to define a core region of the potentials at distances where the corresponding RDFs are zero,
to avoid MC steps leading to such distances. Default: 1000.
- Input-Output parameters:
- Output*
- [BASEOUTFILENAME] Prefix name of the system (filename template) to use for writing
output files. All names of output files will begin with the given prefix.
- VerboseLevel
- [IPRINT] Verbosity level of the log-file. 1-minimum level, 10 - maximum level.
Default: 5.
- WriteTraj
- [ITR] How often (in terms of MC steps) to write current geometry to the trajectory
file. Default: 0 - do not write it at all.
- InputRDF
- [FILRDF] Input file with reference distribution functions. Required for inverse
procedure, otherwise only a direct MC simulation will be performed
- InputPot
- [FILPOT] Input file with a set of trial potentials.
- InputStartCoords
- [FSTART] Name of the input file (or prefix for a set of files) with starting
coordinates, excluding 'p001.start.xmol' suffix. If followed by additional parameter N, then the starting coordinates will be randomly choosen among first N-frames of the file. Usefull when one wants to randlomly start from some preequlibrated geometry. If InputStartCoords is not provided, the starting geometry will be generated randomly.
- InputFrozenCoords
- [FCRD] Name of a single *.xmol file with starting coordinates of all
frozen molecules in the system. It will be used to define starting location of the frozen species
for every inverse iteration. The file is almost the same as start.xmol, but it does not contains
moving molecules/atoms.
- DumpLastConf
- [LXMOL] If true, program dumps the last configuration of MC process in file
(or set of files) with ".start.xmol" extention. It is done after every inverse iteration on every
parallel process. Default: False - do not dump. In case of parallel execution, output filenames
have extentions
<Output>.i<iteration>.p<process>.start.xmol
Next: Example: magic.inp
Up: MagiC core: Inverse Solver
Previous: Input/Output files
Contents
Alexander Lyubartsev
2017-02-03