PotsExport2Gromacs(pots, npoints=2500, Umax=6000,
Rmaxtable=2.5, PHImaxtable=180, filename='', noplot=False, hardcopy=True, figsize=(14, 7.5), dpi=120, sigma=0.5,
zeroforce=True, interpol=True, ofilename='')
This procedure exports a set of potentials into GROMACS's .xvg format. Such an export is a rather advanced and multistage process, here a detailed description of the procedure is given.
The first thing to take into account is that Gromacs requires typically a higher resolution of the grid for tabulated potentials than typical resolution of the RDF inversion which is used in MagiC. Furthermore, the potential and force should be smooth functions of the distance, and do not have discontinuities at the end of the intervals where they are determined. Thus the original range where the potentials and RDFs were determined during the inversion procedure, should be extended in a smooth fashion to nearby range of distances.
Thus in general the question is the following: we start with a tabulated potential which is defined
on a r-grid
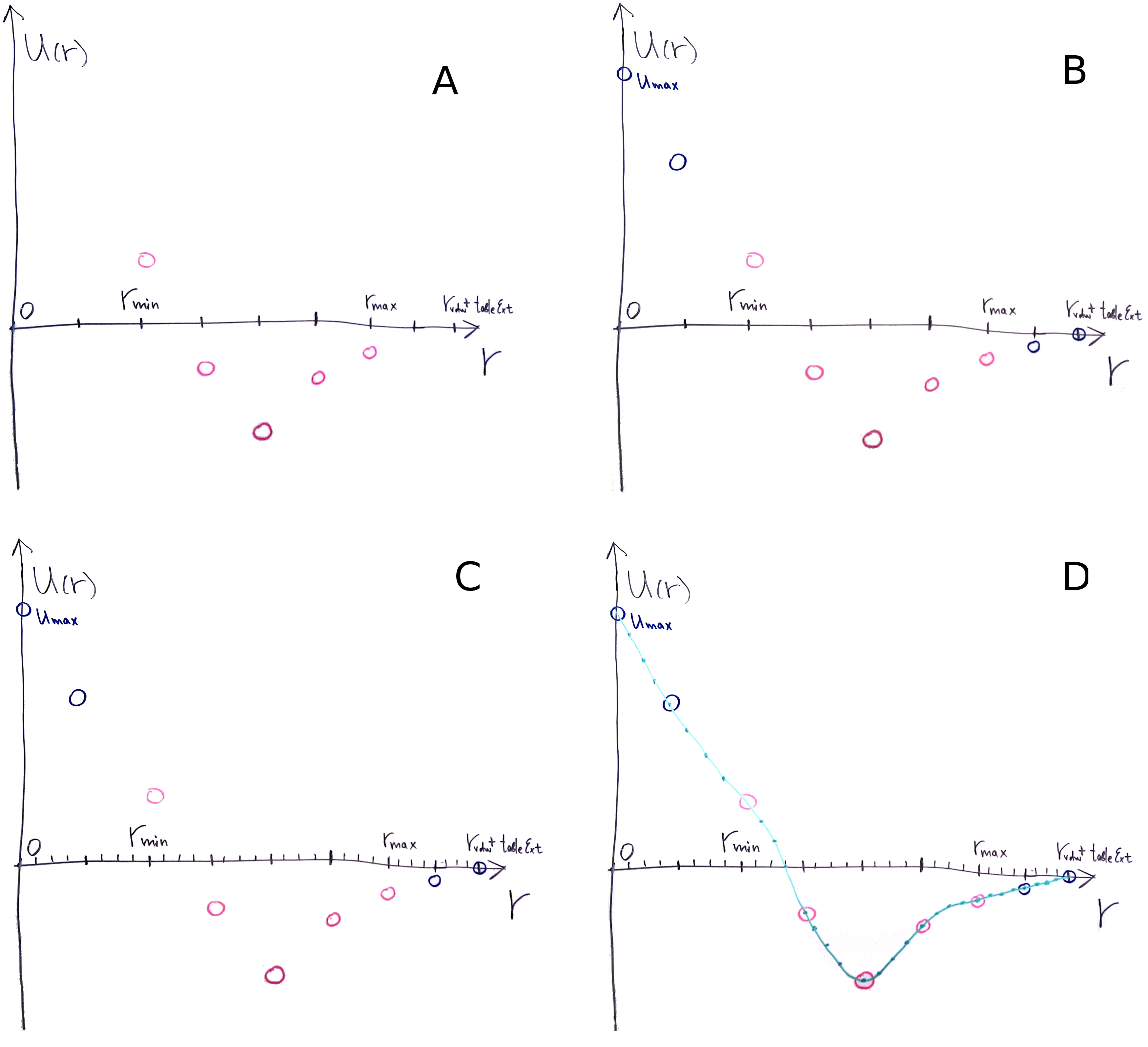
![]() with a given density (figure 4-A, red circles);
and as the result we need to obtain a tabulated potential, which is defined on a r-grid
with a given density (figure 4-A, red circles);
and as the result we need to obtain a tabulated potential, which is defined on a r-grid
![]() with larger density (figure 4-D, cyan line).
with larger density (figure 4-D, cyan line).
In order to do that, we need to introduce left-side and right-side extensions of the potential which should be smoothly connected to the original potential (figure 4-B, blue circles). The left side extension should represent strongly repulsive core so it is approximated by
| (7) |
and coefficients ![]() and
and ![]() are chosen to provide continuity of
are chosen to provide continuity of
![]() at
at ![]() .
For the right side extension we should take into account the origin of the potential: Short-range
intermolecular potentials should decay to zero when
.
For the right side extension we should take into account the origin of the potential: Short-range
intermolecular potentials should decay to zero when ![]() :
:
![$\displaystyle U^{right}_{short\,range}(r)=U(r_{max})\cdot\exp[{-10\frac{(r-r_{max})}{r_{vdw+table-extension}-r_{max}}}]$](img39.png) |
(8) |
![$\displaystyle U^{right}_{angle}(\phi)=U(\phi_{max})\cdot\exp[{-100\frac{(\phi-\phi_{max})}{180^{\circ}-\phi_{max}}}]$](img42.png) |
(9) |
| (10) |
Once the extension has been made, we can interpolate all the points to a denser grid
(figure 4-C). Number of nodes in the grid is defined by npoints.
If interpol=False, a grid of original density is used. The interpolation is made by
Gaussian smoothing, e.g. every point of the resulting potential is obtained as
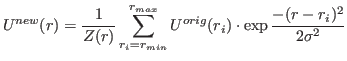 |
(11) |
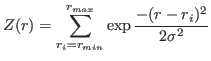 |
(12) |
Once the interpolation is done, each resulting potential and force based on it are written to a xvg-file.
The xvg-file is named by the name and type of the corresponding original potential. Forces can be
suppressed by setting noforce=True, then zeros will be written to the file. In such case
GROMACS should automatically calculate forces from a given potential.
In order to control the results, each original potential, the extrapolated part and the interpolated
potential are plotted together. The plotting is controlled by parameters
noplot, hardcopy, figsize, dpi, which are explained below.
zeroforce=True force values are plotted.
npoints value will be ignored (optional argument).
Default - True, potentials are interpolated.
MagicTools.PotsExport2Gromacs(Pots) - simplest call with default parameters.
MagicTools.PotsExport2Gromacs(Pots[0:10], sigma=0.1, zeroforce=True, interpol=False ) -
export only first 10 potentials from the set; do not use dense net for interpolation;
use averaging with sigma=0.1 (very narrow gaussian); do not export forces, write zeros instead.