Next: Compilation Up: The main input file Previous: Path Integral mode Contents
Output <number>
At output level 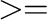 5, type line(s) with some data (time step, energies, etc)
each
5, type line(s) with some data (time step, energies, etc)
each <number> of steps
Default: 1
Average_freq <number>
Defines how often to collect averages: various internal degrees of freedom, partial energies, probabilities of subensembles in the expanded ensemble mode.
Default: 1
Serie_average <number>
Compute and remember intermediate averages over series of <number> steps
Default: 10000
Average_from <number>
Begin final averaging over series (see Serie_average keyword) from
series with the given <number>
Average_internal yes/no
Whether to compute average values of all bond lengths, angles and their energies
Default: no
Dump_XMOL yes/no
Dump the final configuration in “XMOL” format
Default: no
Trajectory <format> <step> <num_conf> <list>
Dump trajectory.
<format> may be one of:
bincrd - binary with coordinates only
binvel - binary with coordinates and velocities (in Å/fs)
asccrd - ACSII (XMOL-format) with coordinates only
ascvel - ACSII (XMOL-type format) with coordinates and velocities
ascfor - ACSII (XMOL-type format) with coordinates and forces (in kJ/mol/Å)
ascfvl - ACSII (XMOL-format) with coordinates, velocities and forces
<step> is time interval (in femtoseconds) between the configurations
saved in the trajectory file
<num_conf> - number of configurations in each trajectory file.
After filling <num_conf> configuration in a trajectory file,
the program begins to write configurations in the next file.
Trajectory files acquire extensions .001, .002, .003, etc
<list> - list (in the form of numbers 0 or 1 for each molecule type)
which signals whether to include molecules of this type into trajectory.
<list> may be set also to “all” (all molecules)
Units for velocities in the trajectory file are Å/fs.
Bond_list yes/no
Generate list of all bonds and put it in file bond.list
Default:no
End
end of the input file. This line is compulsory