Next: Molecular geometry and non-bonded Up: Description of the molecular Previous: Description of the molecular Contents
The general form of the force field implemented in the program is:
where
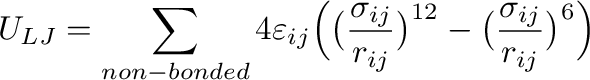 (2)
(2)
is the sum of Lennard-Jones interactions taken over non-bonded atom
pairs. A pair of atoms is considered as non-bonded if they are on
different molecules or if they are on the same molecule but separated by more
than two covalent bonds. A case when a pair of atoms is separated by exactly
3 bonds (the so-called 1-4 neighbors) is treated separately, see below.
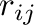 is the distance between atoms
is the distance between atoms 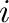 and
and 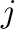 . Parameters
. Parameters
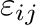 and
and 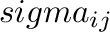 for atom pair (
for atom pair (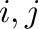 ) are determined
by default from the individual
) are determined
by default from the individual 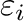 and
and 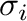 parameters
for atoms using combination rules; they can be also specified separately.
parameters
for atoms using combination rules; they can be also specified separately.
 (3)
(3)
is the sum of electrostatic interactions
The remaining terms in (1) are inramolecular interactions due to covalent bonds, covalent angles and torsion angles. They are described below.
For each molecule type used in the simulation a file describing molecular
structure and parameters of the force field is needed. The
file must have extension *.mmol . Some examples of .mmol files are given
in moldb directory.
.mmol files consist of several parts which are described below.
Lines, beginning with "#" are commentaries and they are ignored by the
program. Other lines contain parameters in free format. The delimiter
between keywords and parameters is one or more spaces. Do not use Tab as
a delimiter, it may result in unpredictable behaviour.
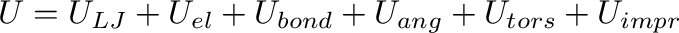 (
(