Next: Ensemble Up: The main input file Previous: Restart control Contents
Molecule_types <num_types> <name> <number> [fixed] ... <name> <number> [fixed]
Which molecules to simulate. <num_types> is the number of molecule
types. This keyword must be followed by <num_types> lines each of
which contains at least two parameters: <name> and <number>
<name> - name of the molecule. File <name>.mmol , describing
the molecular structure and the force field parameters, must be
present in directory specified by parameter Path_DB.
<number> - number of molecules of this type.
“fixed” - Option “fixed” signals that these molecules are fixed and will not move in the MD procedure.
This keyword and its parameters (except “fixed”) are compulsory
PBC <type>
Type of periodic boundary conditions
<type> may be:
rect - rectangular (default)
hexa - hexagonal
octa - truncated octahedron
Box <box-x> <box-y> <box-z>
Specifies simulation box size (in Å). In case of hexagonal boundary
conditions, only box-x and box-z have a meaning.
For octahedral boundary conditions only <box-x> has a meaning.
The keyword cannot be used simultaneously with “Density”
Density <value>
Setup initial density. <value> is density in 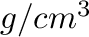 . This option
cannot be used simultaneously with setting up box sizes. In case of
rectangular periodic boundary conditions, a cubic cell is implied. Zero
density implies “vacuum simulation” regime with the box size automatically
set to 1000 Å
. This option
cannot be used simultaneously with setting up box sizes. In case of
rectangular periodic boundary conditions, a cubic cell is implied. Zero
density implies “vacuum simulation” regime with the box size automatically
set to 1000 Å
Combination_rule <type>
Use another (than Lorentz-Berthelot) combination rule for Lennard-Jones parameters for different types
type may be one of the following:
Sigma_geom
LJ 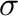 parameter is calculated as a geometrical
average of the both types (as for example in the OPLS force field)
parameter is calculated as a geometrical
average of the both types (as for example in the OPLS force field)
Kong
use Kong rules (J.Kong, J.Chem.Phys., 59, 2464, (1973))
Fender
use Fender-Halsey rules ( B. E. F. Fender and G. D. Halsey, Jr. "Second Virial Coefficients of Argon, Krypton, and Argon-Krypton Mixtures at Low Temperatures", J. Chem. Phys. 1881 (1962))
Waldman
use Waldman-Hagler rules (M. Waldman and A. T. Hagler
"New combining rules for rare-gas Van der-Waals parameters",
Journal of Computational Chemistry 14 pp. 1077-1084 (1993))
Mixed
use mixed combination rules from paper: A. Nikitin, Y. Milchevskiy, A. P. Lyubartsev "AMBER-ii: New Combining Rules and Force Field for Perfluoroalkanes"
J. Phys. Chem. B, 119, 14563-14573 (2015)
Default: use Lorentz-Berthelot combination rules
Pair_list [FF_file] <file_name>
Introduce special Lennard-Jones parameters for pairs of atoms given in
file <filename>.
Without FF_file option the list of pairs is directly composed
from the site numbers given in file <filename>. Each non-commentary
line of this file consists of two integer numbers specifying the global
site numbers of two atoms, followed by two real numbers having the sence of
LJ parameters and 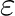 for the given pair of sites,
and eventually followed by two other real numbers which are
LJ parameters and for 1-4 bonded interactions.
This list overwrites the Lennard-Jones parameters defined by the combination
rules and eventually modified by parameters in the
for the given pair of sites,
and eventually followed by two other real numbers which are
LJ parameters and for 1-4 bonded interactions.
This list overwrites the Lennard-Jones parameters defined by the combination
rules and eventually modified by parameters in the .mmol files.
If option FF_file is given, the list of pairs is composed from
the LJ_PAIRS section of the force field file given by parameter
<filename> (same force field file as used by the makemol
utility). The pairs of atoms with force field atom types (given by the
9-th colomn of the .mmol files) which match atom types in the
LJ_PAIRS section of the force field file are included into the list of
pairs for special Lennard-Jones interactions.
El_field <ampl> <freq>
Put the system in a homogeneous time-dependent electrostatic field
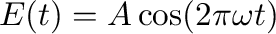 (11)
(11)
where <ampl>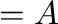 is given in
is given in 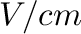 and
and <freq> 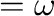 in
in 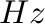 .
.
G-force <type> <g-force>
A gravitation-like force (acting along Z-axis on each atom proportionally
to its mass) is added for molecules of type <type>. Parameter
<g-force gives the value of acceleration 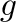 in
in 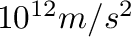 .
The force can be applyed for several molecular types, is this case the keyword
should be repeated for each type.
.
The force can be applyed for several molecular types, is this case the keyword
should be repeated for each type.