Next: Computation of the electron Up: Tranal utility Previous: General organization Contents
This part is common for all types of trajectory analysis. It reads the first part of the input file and then reads trajectories accordingly. The first part of the input file is written in “NAMELIST” format which looks like:
$TRAJ parameter=value(s), ... $END
“TRAJ” is the name of this NAMELIST section.
The following parameters must be defined:
NFORM = <format>
where <format> is one of:
MDYN - MDynaMix binary trajectory (default)
XMOL - XMOL trajectory. It is implied, that the commentary
(second) line of each configuration is written in the format:
(char) <time> (char-s) BOX: <box_x> <box_y> <box_z>
where (char) is any character word, <time> is time in 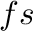 ,
,
<box_x> <box_y> <box_z> (following after keyword BOX) are
the box sizes.
PDBT - PDB trajectory as generated by “trajconv” utility
of GROMACS simulation package
DCDT - DCD trajectories generated by NAMD package
FNAME = <file_name>
In case of multiple trajectory files, set the base name of the trajectory
files. The trajectory must be then written as a sequence of files
<file_name>.001 , <file_name>.002 and
so on, the largest possible number being <file_name>.999 .
In case of a single trajectory file (indicated by parameter NFBEG = -1)
set the full file name (with extension)
PATHDB = <value>
Directory with molecular description files (.mmol). Default is the current directory (.) .
NTYPES = <value>
Number of molecule types in the trajectory
NAMOL = <name1> [,<name2>,...]
NTYPES names of molecules. It is supposed that files
<name1>.mmol, <name2>.mmol,... , or alternatively
<name1>.itp, <name2>.itp,... describing the molecules are
present in the directory defined by PATHDB. Format of .mmol files is
the same as for MDynaMix. Format of .itp files is the same
as in Gromacs, with exception that no “include” statements is allowed.
Tranal looks first for .mmol files, than for .itp files. In case of absence
of any of them, the program still can proceed with the following reservations:
i) correct parameters NSPEC and NSITS are given for all
molecules in the trajectory ii) the masses of all atoms are set to
1, the charges to 0, and atom nanes are undetermined.
NSPEC = <n1>[,<n2>,...]
Number of molecules of each type ( NTYPES numbers). This parameter
is not necessary in MDynaMix binary trajectories.
NSITS = <n1>[,<n2>,...]
Number of atoms on each molecule of each type ( NTYPES numbers).
This parameter is not necessary if .mmol or .itp files for each molecular
type are provided.
NFBEG = <value>
Number of the first trajectory file (integer between 0 and 999). If this
number is negative, a single trajectory file with name given by
parameter FNAME is used.
NFEND = <value>
Number of the last trajectory file (integer between 0 and 999)
IPRINT = <value>
Defines how much you see in the intermediate output. The final output with analysis of results does not depend on it. Default value is 5.
BOXL = <x-box-size> BOYL = <y-box-size> BOZL = <z-box-size>define the box size if it is not present in the trajectory (implies constant-volume simulation)
BREAKM = <value>
Defines which break in trajectory (in ps) is allowed to consider the trajectory as continuous. The parameter is used in analysis of time-dependent properties.
LVEL = <logical>
Accept a logical value. .true. signals that the trajectory
may contain velocities, which will then be read for the analysis.
LXMOL = <logical>
Accept a logical value. If .true., rewrite trajectory in XMOL format
FXMOL = <name>
File name for output XMOL trajectory
ISTEP = <value>
Specifies that only each ISTEP-th configuration from the trajectory
is taken for the analysis
LBOND = <logical>
Accept logical value. If .true., information about bonds is read from .mmol files
VFAC = <value>
Multiply velocities from the trajectory by the given <value>. This
can be used for trajectories prepared by other MD programs than MDynaMix,
in order to bring velocities to Å/fs (the units which are used in the
analysis). In MDynaMix trajectories, velocities are already in Å/fs.
Default is 1.