Next: Radial distribution functions: rdf.f Up: Tranal utility Previous: Lateral diffusion: latdiff.f Contents
This utility computes the order parameters of selected molecular vectors relative to the Z-axis, as well as angular distributions of these vectors.
Input parameters for this utility follow after the trajectory parameters in the following format:
If this parameter is zero (or negative), angular distributions of the specified vectors are computed relative to the positive direction of the Z axis. If this parameter is positive, it defines the type of “lipid” molecules in membrane-like systems. The Z-coordinate of the center of mass of these molecules defines the membrane middle plane, and angular distributions of the molecules which are below the middle plane are calculated relative to the negative direction of the Z-axis.
One line per each order parameter (their number is defined above) Each line can be in one of the following form:
<n1> <n2> 0
Here <n1> and <n2> are the site numbers (must belong to the
same type of molecules) which define the molecular vector. Computation for
this vector is done independently from others
<n1> <n2> 1 <nref1> <nref2>
followed immediately by
<n3> <n4>
Order parameters and angular distributions for molecular vectors
determined by a pair of such lines
are computed together. A scalar product of vectors defined by
<n1> <n2> and <nref1> <nref2> is compared with scalar
product defined by <n3> <n4> and <nref1> <nref2>. If the
first scalar product is greater, then the vector <n1> <n2> is
considered as going first and the vector <n3> <n4> next; if
the second scalar product is greater, then the order in which these vectors
follow is changed to opposite (as these lines change places). Note that
this comparison is made for each molecule separately. Such arrangement has a
sense if, for instance, two hydrogens, equivalent from the force field point
of view, are distinguishable in each specific configuration by relation to
another, “reference” vector, defined by numbers <nref1>,<nref2>,
like so-called G1R and G1S order parameters in the G1 glycerol atom of
phospholipids.
The last line is an integer. If 0, no orientational distributions are
computed. If non-zero, it defined the number of “bins” into which
the interval ![$[-1,1]$](img90.png) is divided for calculation of
is divided for calculation of 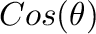 distribution.
distribution.
further comments.
The order parameter for a specific molecular vector is defined as
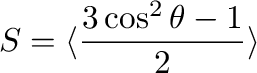
where 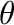 is the angle between the molecular vector and Z-axis, and
averaging is taken over all molecules and time frames.
The output lists also NMR dipole coupling for the corresponding vectors as:
is the angle between the molecular vector and Z-axis, and
averaging is taken over all molecules and time frames.
The output lists also NMR dipole coupling for the corresponding vectors as:
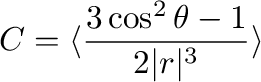
where  is the length of the corresponding molecular vector.
is the length of the corresponding molecular vector.