PATH INTEGRAL MONTE CARLO METHOD
The method is based on Feynman path integral interpretation of Quantum
mechanics. Each quantum particle is represented by a "ring polymer" whose
elements (beads) are connected by harmonic forces ("springs").
Interparticle interactions are expressed as interactions between simultaneous
beads of the polymers.
The Path Integral Monte Carlo (PIMC) method provides a direct, non-shifted
statistical evaluation of quantum canonical averages:
< A > = Tr(A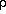 )
)
where =
exp(-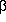 H)
is the canonical density matrix, H is the
Hamiltonian and is
the reciprocal temperature.
H)
is the canonical density matrix, H is the
Hamiltonian and is
the reciprocal temperature.
The great advantage of the PIMC method is that it allows, in principle,
really ab initio, without any assumptions beyound the Schrödinger
equation, simulations of quantum systems. Still, an application of the
PIMC method to a real system of quantum particles
(e.g. electrons in the field of nuclei) at finite temperature
remains a challenging problem. The main obstacles, both of principal and
computational nature, come from the necessity of
antisymmetrisation of the density matrix for a many-electron system.
The following paper provides a methodological basis for application
of the PIMC method to the system of quantum identical particles with spin 1/2.
A.P.Lyubartsev, P.N.Vorontsov-Velyaminov.
"Path Intergal Monte Carlo method in quantum statistics
for a system of N identical fermions"
Physical Review A, v.48(6), pp.4075-4083 (1993)
Abstract
A rigorous expression for the partition function of N identical
fermions with spin 1/2 is obtained in a form suitable for Path Integral
Monte Carlo (PIMC) simulation of a many electron system at finite
temperature. A sum over permutations is reduced to a sum over
classes with readily determinable factors. Distribution over
projections and values of the total spin is also obtained enabling PIMC
calculations of spin-dependent quantities. A new MC algorithm is
proposed which accounts simultaneously for all types of trajectory
linkages. A test calculation - four electrons in a spherical cavity with
radius 1 nm at temperature 300 - 2000 K is performed.
See also:
P.N.Vorontsov-Velyaminov, M.O.Nesvit and R.I.Gorbunov, "Bead-Fourier
path integral Monte Carlo method applied to systems of identical
particles", Rhys.Rev.E, v.55(2),p.1979-1997 (1997)
Recent papers on the subject:
P.N.Vorontsov-Velyaminov and A.P.Lyubartsev, "Entropic sampling
in the Path Integral Monte Carlo method", J. Phys. A: Math. Gen.
v. 36, pp. 685-693 (2003)
Abstract; Text (PDF).
This paper describes application of Entropy sampling approach to compute
density of states and thermodynamic averages of a quantum system
- S.D.Ivanov, A.P.Lyubartsev and A.Laaksonen
"Bead-Fourier Path Integral Molecular Dynamics"
Phys. Rev. E, v.67, 066710 (2003)
Full text (APS)
Abstract
Molecular dynamics formulation of Bead-Fourier path integral
method for simulation of quantum systems at finite temperatures is
presented. Within this scheme, both the bead coordinates and Fourier
coefficients, defining the path representing the quantum particle,
are treated as generalized
coordinates with corresponding generalized momenta and masses.
Introduction of the Fourier harmonics together with the center-of-mass
thermostating scheme is shown to remove the ergodicity problem,
known to pose serious difficulties in standard path integral
molecular dynamics simulations. The method is tested for quantum
harmonic oscillator and hydrogen atom (Coulombic potential).
The simulation results are compared with the exact analytical
solutions available for these both systems. Convergence of the
results with respect to the number of beads and Fourier harmonics
is analyzed. It was shown that addition of a few Fourier harmonics
already improves the simulation results substantially, even for
a relatively small number of beads. The proposed Bead-Fourier
Path-Integral molecular dynamics is a reliable and efficient
alternative to simulations of quantum systems.