Next: Startup Up: The main input file Previous: Ensemble Contents
Time_step <value>
Time step in femtoseconds. This is the long time step in case of the double time step algorithm.
Default: 2 fs.
Number_steps <number>
How many MD steps to run.
Double_timestep <number>
Double time step algorithm by Tuckerman et al. <number> is the number
of short time steps in one long. The keyword cannot be used simultaneously
with Constrain.
Constrain <toler> <i_1> <i_2> <i_3> .... <i_ntypes>
Use SHAKE algorithm to constrain the bond lengths. The option cannot be used
with Double_timestep. <toler> is the tolerance level.
<i_1> <i_2> ... <i_ntypes> are numbers 0 or 1, which specify
whether (1) or not (0) to apply constraints to molecules of each species.
R_cutoff <value>
Cut-off (in Å) for the Lennard-Jones and real-space part of the electrostatic forces.
Default: 12 Å
R_short <value>
Cut-off (in Å) for Lennard-Jones forces computed each short-time step (has an effect only in double-time step algorithm)
Default: 5 Å
Neighbour_list <number>
Update the list of neighbors (Verlet list) every <number> steps
Default: 10
Electrostatics <type> [<A> <B>]
How to treat electrostatics
<type> may be Ewald (default), RF (reaction field) and Cutoff
For Ewald: A is
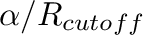 , where
, where 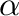 is the Ewald convergence
parameter. Precision of the real-space Ewald part is determined by
is the Ewald convergence
parameter. Precision of the real-space Ewald part is determined by 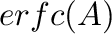 .
.
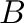 defines the number of terms in the reciprocal part. It cuts the reciprocal
series when expression in the exp of the reciprocal part exceeds .
Recommended values A = 2.5 – 3; B = 7 – 10.
defines the number of terms in the reciprocal part. It cuts the reciprocal
series when expression in the exp of the reciprocal part exceeds .
Recommended values A = 2.5 – 3; B = 7 – 10.
For reaction field, A is the dielectric permittivity and B is the Debye screening length in Å. Setting the Debye length to 0 means an infinite Debye length, i.e non-conducting solution.
If <type> is “Cutoff”, parameters 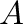 and are not necessary, and
no special treatment of electrostatic forces out of
and are not necessary, and
no special treatment of electrostatic forces out of 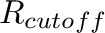 takes place.
takes place.
Default: Ewald method with A=2.8 and B=9.
Cut_forces <value>
If this option is specified and the absolute force acting on any atom
exceeds the specified <value>, the force will be cut to this level while
maintaining the direction. The value is given in Å and has a sense
of the maximum allowed additional displacement during one time step
( F*dt**2/m ) caused by this force
Default: Do not cut forces
Bind_atoms <file> <deviation>
If this keyword is specified, atoms defined in file <file> will be
bound to corresponding positions (given in the same file) by a
harmonic potential with characteristic deviation given by <deviation>
Control_temp <factor>
Activate control of temperature during the simulation. If instant temperature
deviates from the target (thermostat) temperature by factor more than
<factor>, the program stops simulation and prints current
configuration of the system
Default: Off