Next: Expanded Ensemble mode Up: The main input file Previous: Molecular Dynamics Contents
Startup <type> [<optional parameters>]
How to set up initial coordinates.
This option has an effect only if Read_restart was specified as “no”
<type> may be one of the following:
xmol (default) - the input configuration is taken from file
<Main_filename>.start
(see keyword “Main_filname”) which should be in “XMOL” format
The second (commentary) line of this file may contain box sizes (which
follow after keyword BOX:). They will be used as actual box sizes
if keyword “Change_V” is not set or set to “no”. If
keyword “Change_V” is set to “yes” or “slowly” , the box sizes
specified by keywords “Box” or “Density” will be used.
xyz - the input configuration is taken from coordinates written as
three columns of X Y Z coordinates in <Main_filename>.start file
Mol_COM - molecular center-of mass coordinates are taken from
<Main_filename>.start file, as three columns of X Y Z coordinates.
FCC - molecular center of mass coordinates are put on a FCC lattice
Cubic - molecular center of mass coordinates are put on a cubic
lattice
Cyl_hole <radius>
Sph_hole <radius>
Molecules, except those defined in option Start_rot are distributed
outside sphere or cylinder of radius <radius>
In all cases, except “xmol” and “xyz”, atom coordinates are build around molecular center-of-mass using local coordinates specified in the corresponding .mmol files.
Start_rot <i_1> <i_2> <i_3> ... <i_ntypes>
Tells how the molecular coordinates defined in .mmol files are treated at startup:
If Startup options “xmol” or “xyz” are specified, keyword
Start_rot do not have any effect.
Startup option “FCC” or “cubic”:
If i_n=0, molecules with coordinates from -mmol file are
rotated randomly
If i_n=1, coordinates of the molecule of this type are taken from .mmol
file without any change.
If i_n=2, coordinates of the molecule of this type are taken from .mmol
and translated without rotation so that the molecular center of mass will
be at zero.
Startup options “Cyl_hole” and “Sph_hole”:
Molecules which should be placed inside cylindrical or spherical hole,
are marked by parameter i_n = 1 or i_n = 2. Initial
coordinates of atoms of such molecules are taken from the corresponding
.mmol file and it can be only a single molecule of this type.
In case i_n = 2 the molecular coordinates are translated so that
center of mass is set to the coordinates origin. Other molecules
(with i_n = 0) are distributed outside the spherical or
cylindrical hole.
Cases i_n 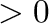 have a sense only if only one molecule of this
type is present.
have a sense only if only one molecule of this
type is present.
Default: all i_n=0 (rotate molecules randomly)
Gather
Gather each molecule in one place (if its atoms were spread over different periodical images). Works both at start-up and at restart.
Default: use coordinates as they are.